There is a common principle that underlies all supervised machine learning algorithms for predictive modeling.
In this post you will discover how machine learning algorithms actually work by understanding the common principle that underlies all algorithms.
Kick-start your project with my new book Master Machine Learning Algorithms, including step-by-step tutorials and the Excel Spreadsheet files for all examples.
Le’s get started.
How Machine Learning Algorithms Work Photo by GotCredit, some rights reserved.
Let’s get started.
Learning a Function
Machine learning algorithms are described as learning a target function (f) that best maps input variables (X) to an output variable (Y).
Y = f(X)
This is a general learning task where we would like to make predictions in the future (Y) given new examples of input variables (X).
We don’t know what the function (f) looks like or it’s form. If we did, we would use it directly and we would not need to learn it from data using machine learning algorithms.
It is harder than you think. There is also error (e) that is independent of the input data (X).
Y = f(X) + e
This error might be error such as not having enough attributes to sufficiently characterize the best mapping from X to Y. This error is called irreducible error because no matter how good we get at estimating the target function (f), we cannot reduce this error.
This is to say, that the problem of learning a function from data is a difficult problem and this is the reason why the field of machine learning and machine learning algorithms exist.
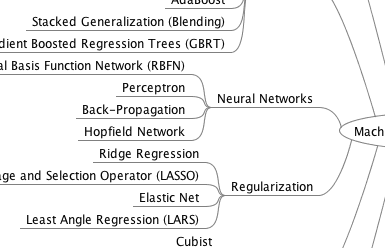
Get your FREE Algorithms Mind Map
Sample of the handy machine learning algorithms mind map.
I've created a handy mind map of 60+ algorithms organized by type.
Download it, print it and use it.
Also get exclusive access to the machine learning algorithms email mini-course.
Learning a Function To Make Predictions
The most common type of machine learning is to learn the mapping Y=f(X) to make predictions of Y for new X.
This is called predictive modeling or predictive analytics and our goal is to make the most accurate predictions possible.
As such, we are not really interested in the shape and form of the function (f) that we are learning, only that it makes accurate predictions.
We could learn the mapping of Y=f(X) to learn more about the relationship in the data and this is called statistical inference. If this were the goal, we would use simpler methods and value understanding the learned model and form of (f) above making accurate predictions.
When we learn a function (f) we are estimating its form from the data that we have available. As such, this estimate will have error. It will not be a perfect estimate for the underlying hypothetical best mapping from Y given X.
Much time in applied machine learning is spent attempting to improve the estimate of the underlying function and in term improve the performance of the predictions made by the model.
Techniques For Learning a Function
Machine learning algorithms are techniques for estimating the target function (f) to predict the output variable (Y) given input variables (X).
Different representations make different assumptions about the form of the function being learned, such as whether it is linear or nonlinear.
Different machine learning algorithms make different assumptions about the shape and structure of the function and how best to optimize a representation to approximate it.
This is why it is so important to try a suite of different algorithms on a machine learning problem, because we cannot know before hand which approach will be best at estimating the structure of the underlying function we are trying to approximate.
Summary
In this post you discovered the underlying principle that explains the objective of all machine learning algorithms for predictive modeling.
You learned that machine learning algorithms work to estimate the mapping function (f) of output variables (Y) given input variables (X), or Y=f(X).
You also learned that different machine learning algorithms make different assumptions about the form of the underlying function. And that when we don’t know much about the form of the target function we must try a suite of different algorithms to see what works best.
Do you have any questions about how machine learning algorithms or this post? Leave a comment and ask your question and I will do my best to answer it.
Is it possible to learn Machine learning without prior guidance? I don’t have enough physical resources like a professor or a expert in Machine learning. I was just interested in learn programming which about prediction and feeding the data into computer to make to predict the circumstances and predict the future to take the right decisions. Well, as normal student having limited resources, is it really possible to dive into Machine learning. What are the prerequisites for Machine learning? What is the best alternative to get a live guidance to take Machine learning? Please help!
Great read!
I am just getting started in Machine Learning. My question after reading is, do the machine learning algorithms try to alter the mapping function f(X) to reduce error, or do they only try to create a mapping function from given data sets of (X,Y)? Or is it both?
Also, what does the mapping function look like? For a standard set of X and Y variables that are floating point numbers, would it be something of the form (Y = mX + b)? More quadratic or even approaching differential equations or linear algebra? Does the mapping function come from trying to make a line of best fit on a graph from a set of data?
Sorry for all my questions. I am eager to learn!
Thank you!
i didnt know about machine learning but i take the college project related to machine learning so i now started to learn machine learning its intresting and very well i love maths i learned python day and night watching tutorials and learn from websites
Sir, as referred to in the article the statistical inference, that is the mathematical relationship between the input data and the predicted values…or the mathematical function…how much of an importance does it have for an ML engineer? Thank you for your help!!!
Good evening, I am a learner wants to start my work in the field of AI.And I have done some part in Soft computing.kindly guide me so that I can start my work as a beginner in the field of AI.
Sir, I need some basic operation of RBF kernel based learning and on Reproducing kernel hilbert spaces (RKHS) using GRAM Matrix along with their MATLAB implementation for my research work in Ph.D. Kindly guide me on above topics.
I am trying to modify your script to create Adaptive Random Forest alghoritm, but I faced many problems.
I created the function which stores examples within window and wait until some part of examples will be stored, then I am trying to use the implemented methods.
Unfortunately I am unable to do that.
Could you give me some advices ? Or some slices of code/pseudocode?
Your posts are just awesome for people having no idea what ML(Machine Learning) is. Stuffs are really good and easily interpretative. That also show the efforts you have put in to master it. I have a query:
Is cloud computing services knowledge like AWS, Azure or GCP required before learning ML.
Hi Jason, Your expertise and knowledge in these articles you write is quite impressive! Thank you for taking the time to share.
My question is this, using machine learning – assuming we find a good model for Y = f(x1, x2, x3)… Once we have established this model, can we use the determined relationship to provide a Y value and have the model estimate x1, x2, x3? I would like to think we could since equations of this sort are generally reversible… What type of machine learning algorithms and methods would you recommend for this sort of problem?
No, the reverse modeling problem is significantly harder. Off the cuff (and probably wrong), it sounds like an optimization problem – find me a set of inputs to achieve the desired output.
I have doubt regarding these statements and find it a bit difficult to draw the line of difference between the 2.
>> The most common type of machine learning is to learn the mapping Y=f(X) to make predictions of Y for new X.
This is called predictive modeling or predictive analytics and our goal is to make the most accurate predictions possible.
>>We could learn the mapping of Y=f(X) to learn more about the relationship in the data and this is called statistical inference. If this were the goal, we would use simpler methods and value understanding the learned model and form of (f) above making accurate predictions.
Statement 1 is purely telling that predictive modeling/predictive analytics is not really bothered about what form function f takes but it concentrates more towards the accuracy of the prediction itself. For example, with the iris data set, post training, how accurate is the function’s output to the actual output. This is what predictive modeling/analytics is concerned about
Statement 2 tells that statistical inference is something that is concerned about the relationship between X and Y and not about the function’s output itself. For example, lets consider that for a dataset that I have which relates an area’s population to its temperature, the inference might be that with increasing population, the overall temperature of an area increases. So these 2 parameters are directly proportional. This inference is what statistical inference is concerned about and not the accuracy with which function f predicts the data.
Is this understanding right? Kindly guide and help me with some examples.
Yes, they are related, and one can be used for the other.
Sometimes understanding the relationship can come at the expense of lower predictive accuracy, e.g. we use a linear model because we can interpret it, instead of a complex ensemble of decision trees that we cannot interpret.
In machine learning, the representational power of a model reflects what target functions are representable by it. Then, it seems that the more representational power a model has, the better is the model. What do you think?
Even if a model has a lot of power, maybe you care more about being able to explain/interpet the model and choose a worse performing and simpler model instead.
Hello, I am trying to download the Free Mind Map on the webpage, and am unable to do it. Any chance you can email me the pdf form please? It opens up the new webpage, but then crashes on me. I’m on Brave, so maybe that has something to do with it.



")

 for Machine Learning")


Is it possible to learn Machine learning without prior guidance? I don’t have enough physical resources like a professor or a expert in Machine learning. I was just interested in learn programming which about prediction and feeding the data into computer to make to predict the circumstances and predict the future to take the right decisions. Well, as normal student having limited resources, is it really possible to dive into Machine learning. What are the prerequisites for Machine learning? What is the best alternative to get a live guidance to take Machine learning? Please help!
Yes, absolutely Sreepal.
Start here:
https://machinelearningmastery.com/how-do-i-get-started-in-machine-learning/
Great read!
I am just getting started in Machine Learning. My question after reading is, do the machine learning algorithms try to alter the mapping function f(X) to reduce error, or do they only try to create a mapping function from given data sets of (X,Y)? Or is it both?
Also, what does the mapping function look like? For a standard set of X and Y variables that are floating point numbers, would it be something of the form (Y = mX + b)? More quadratic or even approaching differential equations or linear algebra? Does the mapping function come from trying to make a line of best fit on a graph from a set of data?
Sorry for all my questions. I am eager to learn!
Thank you!
Depends on the algorithm, often algorithms seek a mapping with min error.
Algorithms like knn have no such optimization or functional form.
How we know the value of error, since we dont know exactly the value of Y?
We don’t and some error will always exist.
i didnt know about machine learning but i take the college project related to machine learning so i now started to learn machine learning its intresting and very well i love maths i learned python day and night watching tutorials and learn from websites
Hang in there!
i am confused …………which algorithm is gives best results in privacy preserving for different data sets…….
It depends on the data. My advice is to test on your data and discover what works best.
Sir, as referred to in the article the statistical inference, that is the mathematical relationship between the input data and the predicted values…or the mathematical function…how much of an importance does it have for an ML engineer? Thank you for your help!!!
A conceptual understanding of this relationship is of the highest importance for getting the most out of a given prediction problem.
It provides a framework for thinking about your problem.
Good evening
if you can help me with the code and schema of algorithm “LSTM” because I need it in my own research in the master certificate
Thank you
I have many examples, start here:
https://machinelearningmastery.com/start-here/#lstm
Good evening, I am a learner wants to start my work in the field of AI.And I have done some part in Soft computing.kindly guide me so that I can start my work as a beginner in the field of AI.
You can start here:
https://machinelearningmastery.com/start-here/#getstarted
Sir, I need some basic operation of RBF kernel based learning and on Reproducing kernel hilbert spaces (RKHS) using GRAM Matrix along with their MATLAB implementation for my research work in Ph.D. Kindly guide me on above topics.
This is a common question that I answer here:
https://machinelearningmastery.com/faq/single-faq/what-research-topic-should-i-work-on
Hello sir,
What is representation in above context?
What is meant by shape and form of function?
Thank You
We don’t know the shape and form of the function, we use algorithms to approximate it by minimizing loss.
If we did know about the function, we would just use it directly and there would be no need to learn anything.
Hello Jason,
I am trying to modify your script to create Adaptive Random Forest alghoritm, but I faced many problems.
I created the function which stores examples within window and wait until some part of examples will be stored, then I am trying to use the implemented methods.
Unfortunately I am unable to do that.
Could you give me some advices ? Or some slices of code/pseudocode?
Perhaps this will help:
https://machinelearningmastery.com/implement-random-forest-scratch-python/
Hi Jason,
Your posts are just awesome for people having no idea what ML(Machine Learning) is. Stuffs are really good and easily interpretative. That also show the efforts you have put in to master it. I have a query:
Is cloud computing services knowledge like AWS, Azure or GCP required before learning ML.
Thanks.
No, you can run most models on in memory datasets on your own workstation.
Hi Jason, Your expertise and knowledge in these articles you write is quite impressive! Thank you for taking the time to share.
My question is this, using machine learning – assuming we find a good model for Y = f(x1, x2, x3)… Once we have established this model, can we use the determined relationship to provide a Y value and have the model estimate x1, x2, x3? I would like to think we could since equations of this sort are generally reversible… What type of machine learning algorithms and methods would you recommend for this sort of problem?
Thanks Nate.
No, the reverse modeling problem is significantly harder. Off the cuff (and probably wrong), it sounds like an optimization problem – find me a set of inputs to achieve the desired output.
At least for non-linear models.
I have doubt regarding these statements and find it a bit difficult to draw the line of difference between the 2.
>> The most common type of machine learning is to learn the mapping Y=f(X) to make predictions of Y for new X.
This is called predictive modeling or predictive analytics and our goal is to make the most accurate predictions possible.
>>We could learn the mapping of Y=f(X) to learn more about the relationship in the data and this is called statistical inference. If this were the goal, we would use simpler methods and value understanding the learned model and form of (f) above making accurate predictions.
Statement 1 is purely telling that predictive modeling/predictive analytics is not really bothered about what form function f takes but it concentrates more towards the accuracy of the prediction itself. For example, with the iris data set, post training, how accurate is the function’s output to the actual output. This is what predictive modeling/analytics is concerned about
Statement 2 tells that statistical inference is something that is concerned about the relationship between X and Y and not about the function’s output itself. For example, lets consider that for a dataset that I have which relates an area’s population to its temperature, the inference might be that with increasing population, the overall temperature of an area increases. So these 2 parameters are directly proportional. This inference is what statistical inference is concerned about and not the accuracy with which function f predicts the data.
Is this understanding right? Kindly guide and help me with some examples.
Yes, they are related, and one can be used for the other.
Sometimes understanding the relationship can come at the expense of lower predictive accuracy, e.g. we use a linear model because we can interpret it, instead of a complex ensemble of decision trees that we cannot interpret.
Hello.
How do we find the function having those dots?
What function are you referring to exactly?
In machine learning, the representational power of a model reflects what target functions are representable by it. Then, it seems that the more representational power a model has, the better is the model. What do you think?
It really depends on the goal of the project.
Even if a model has a lot of power, maybe you care more about being able to explain/interpet the model and choose a worse performing and simpler model instead.
Merci beaucoup pour vos tutoriels
You’re very welcome!
I really need to learn what happens when it comes to calculating the error function in the context of deep learning
Perhaps start here:
https://machinelearningmastery.com/how-to-choose-loss-functions-when-training-deep-learning-neural-networks/
Hello, I am trying to download the Free Mind Map on the webpage, and am unable to do it. Any chance you can email me the pdf form please? It opens up the new webpage, but then crashes on me. I’m on Brave, so maybe that has something to do with it.
It should send on email. Was that went to your spam folder?